12.15.2. Run FS on a cluster (NIH’s Biowulf) with AFNI¶
Introduction¶
Download script: fs_biowulf.tcsh
Overview¶
We assume you have read the general notes on
running FreeSurfer (FS; specifically recon-all) and AFNI together.
For those of us using NIH’s Biowulf cluster to run FS, there are some
extra considerations, conveniences and caveats. We cover some of
those here and provide some recommendations. At the bottom of this
page, we provide a script for running FS’s recon-all and the AFNI
converter @SUMA_Make_Spec_FS completely, just specifying the input
dset, the subject ID and a couple relevant path names.
We focus on tcsh scripting here, because those are what we mostly
use (for syntax convenience and relative readability).
For reference:
here is the main Biowulf User Guide
and here is a handy webpage about the FreeSurfer module on Biowulf
Note
You can check the jobs you have submitted on Biowulf with:
sjobs -u USERNAME
... for example to see if your job is running or just queueing, and to get your job ID number.
Module loading FS (and other things) in a script¶
When using Biowulf, there a couple things to know about loading modules in a script, esp. if you are using FreeSurfer.
If all three of the following are true for your use usage case:
your terminal shell is
bash(i.e., ifecho $0shows something with “bash” in it)your script is
tcshorcsh(i.e., the shebang at the top is#!/bin/tcshor#!/usr/bin/env tcshor similar)your script loads one or more modules (i.e., it contains
module load ...)
... then you need to include the following line before loading the module(s):
source /etc/profile.d/modules.cshThis is described more in the Using modules in scripts section here, if you don’t believe me.
When using FreeSurfer, you also need to source the magical setup file for the particular shell. For a
tcshscript, you need to include the following line after loading the module(s):source $FREESURFER_HOME/SetUpFreeSurfer.csh
... where
$FREESURFER_HOMEshould be a known variable within the shell once you have loaded the FreeSurfer module, so you don’t need to worry about defining it yourself.
Thus, when I have a tcsh script to run FreeSurfer and AFNI
(and yes, I do use bash as my login shell), I include the
following lines at the very top (just after the shebang):
source /etc/profile.d/modules.csh
module load afni freesurfer
source $FREESURFER_HOME/SetUpFreeSurfer.csh
And you can, too!
Using scratch disks as temporary directories¶
The read/write I/O in the main Biowulf directories where you likely work and save things can be pretty slow. It is actually better to work on temporarily-allocated “scratch” disks and then copy everything back to where you actually want it in your own file tree.
These scratch space allocations are made when you submit your script
to run using sbatch, by including an option like
--gres=lscratch:10. This would give me a 10GB directory on the
scratch disk, in the toplevel directory called
/lscratch/$SLURM_JOBID, where $SLURM_JOBID is provided by the
job allocation system. We can write data here or make a subdirectory
to write into, for example /lscratch/$SLURM_JOBID/SUBJ_ID, where
SUBJ_ID would be the subject ID.
The way we typically include this in a script is to do the following:
Define a directory we want to output to within our real directory structure.
Then check if we have asked for+been allocated space on the scratch disk for temporary I/O, by asking if a directory
/lscratch/$SLURM_JOBIDexists. This will be true if we have submitted our job to the slurm system withsbatchorslurmwith a particular--gres=lscratch:VALUEoption, whereVALUEshould be replaced with amount of desired disk space, in units of GB.If the directory does exist, we can write our outputs there first, and then later copy everything back (more efficient when we have large jobs that read/write a lot of data)
If it does not exist, we will just output to our desired location directly (the less efficient way, likely, for larger work)
The chunk of code to do this could look like:
set subj = sub-001
set dir_fs = ./group_analysis_dir
# Set temporary output directory; then requires using something like
# this on the swarm command line: --sbatch '--gres=lscratch:10'.
# These variables used again *after* the main commands, if running
# on Biowulf.
if ( -d /lscratch/$SLURM_JOBID ) then
set tempdir = /lscratch/$SLURM_JOBID
set usetemp = 1
else
set tempdir = ${dir_fs}
set usetemp = 0
endif
Note
Just checking if $SLURM_JOBID is defined is not a good
enough condition here. Starting a sinteractive or
spersist node is actually a slurm job itself, so the
$SLURM_JOBID variable will be defined even then, and
that doesn’t mean we have space allocated on the scratch
disk. Though, we could start the session with a
--gres=lscratch:VALUE option, and in that case make use
of temporary scratch disk space then.
Then, after we have done our work, we can see if we need to copy
everything back. If we don’t, all our data should be in the right
spot already (${dir_fs}, above). If we do, it is just a matter of
doing the copy. The following if condition covers all we should
need:
# Again, Biowulf-running considerations: if processing went fine and
# we were using a temporary directory, copy data back.
if( $usetemp && -d ${tempdir} ) then
echo "++ Copy from: ${tempdir}"
echo " to: ${dir_fs}"
\mkdir -p ${dir_fs}
\cp -pr ${tempdir}/${subj} ${dir_fs}/.
endif
A general biowulf script¶
Let’s take what we have learned above and create a full script to run
FS’s recon-all and AFNI’s @SUMA_Make_Spec_FS. The script will
be broad enough to be submitted using sbatch or slurm (for
using the scratch disk as a temporary dir), or just with tcsh ..
(which might be a good deal slower, due to disk I/O).
We control the allocated memory, number of CPUs, and scratch disk
space when we submit the job with sbatch. Here, I intend to use
the -parallel option in recon-all (see timing results
here), so I will allocate 4 CPUs to use
(though note that the program can sometimes crash using this option;
see the end of this section of the notes for more on the error/crash). In the
script, the if ( $?SLURM_CPUS_PER_TASK ) ... conditional can then
set our OMP_NUM_THREADS value to match this; if we weren’t using
sbatch to submit the job, this variable simply wouldn’t exist, and
no harm is done.
We put a comment in the top of the script for one way to run this
script with sbatch. At present, the “norm” partition nodes seem
like a good ones to use. I am not certain if the “quick” partition
(which maxes out at 4 hours of usage) gives us enough time to reliably
finish a recon-all run, even with the -parallel option. If
you find it does, then you can add that as a partition option via
--partition=norm,quick.
You should also set the amount of time for allocated running. The
default is 2 hrs. I am setting it here for 12 hrs: with
-parallel, I really hope it doesn’t take this long. Requesting too
long of a run time allocation might mean you have to wait longer for
resources to come your way to actually run (the same is true of
overestimating the amount of scratch disk space you need; 10 GB could
likely be reduced for FS with 1 mm isotropic voxels, but the data only
gets gzipped at the end of its run).
Note
If you forget to set the walltime properly or decide later
you need more, then you are in luck! Biowulf has a nice
feature whereby you can expand (or reduce) the walltime on
the fly, using the newwall command. To change a job
with job ID 12345 to run for 12 hours, run the following in
the terminal:
newwall --jobid 12345 --time 12:00:00
Terminal text will tell you if you are successful. After a few seconds or so, you can also verify the update by checking your resources with:
sjobs -u USERNAME
To run this script, you just need to provide 4 pieces of information at the top:
input |
variable meaning/description |
|---|---|
|
input anatomical dset (likely a raw, T1w volume) |
|
subject ID; will be used for both FS output directory name and later volume/surface file names |
|
top level directory for FS output: can be relative or absolute path, and does not need to exist already (will be created on the fly); the actual output directory for this subject will be ${dir_fs}/{subj} |
|
directory where text output monitoring/recording the terminal output will go |
Here is the script:
#!/bin/tcsh
### Run this command with something like
#
# sbatch \
# --partition=norm \
# --cpus-per-task=4 \
# --mem=4g \
# --time=12:00:00 \
# --gres=lscratch:10 \
# do_*.tcsh
#
# ===================================================================
source /etc/profile.d/modules.csh
module load afni freesurfer
source $FREESURFER_HOME/SetUpFreeSurfer.csh
set dset = anat_02_anon.reface.nii.gz
set subj = sub_02
set dir_fs = group_fs
set dir_echo = .
# ---------------- Biowulf slurm check and initializing ----------------
# Set thread count if we are running SLURM
if ( $?SLURM_CPUS_PER_TASK ) then
setenv OMP_NUM_THREADS $SLURM_CPUS_PER_TASK
endif
# Set temporary output directory; then requires using something like
# this on the swarm command line: --sbatch '--gres=lscratch:50'.
# These variables used again *after* the main commands, if running
# on Biowulf.
if ( -d /lscratch/$SLURM_JOBID ) then
set tempdir = /lscratch/$SLURM_JOBID
set usetemp = 1
else
set tempdir = ${dir_fs}
set usetemp = 0
endif
\mkdir -p ${tempdir}
# record any failures; def: no errors
set EXIT_CODE = 0
# ---------------------- Run programs of interest ----------------------
set nomp = `afni_check_omp`
echo "++ Should be using this many threads: ${nomp}" \
> ${dir_echo}/o.00_fs_${subj}.txt
time recon-all \
-all \
-3T \
-sd ${tempdir} \
-subjid ${subj} \
-i ${dset} \
-parallel \
|& tee -a ${dir_echo}/o.00_fs_${subj}.txt
if ( $status ) then
echo "** ERROR running FS recon-all" \
|& tee -a ${dir_echo}/o.00_fs_${subj}.txt
set EXIT_CODE = 1
goto JUMP_EXIT
else
echo "++ GOOD run of FS recon-all" \
|& tee -a ${dir_echo}/o.00_fs_${subj}.txt
endif
@SUMA_Make_Spec_FS \
-fs_setup \
-NIFTI \
-sid ${subj} \
-fspath ${tempdir}/${subj} \
|& tee ${dir_echo}/o.01_suma_makespec_${subj}.txt
if ( $status ) then
echo "** ERROR running @SUMA_Make_Spec_FS" \
|& tee -a ${dir_echo}/o.01_suma_makespec_${subj}.txt
set EXIT_CODE = 2
goto JUMP_EXIT
else
echo "++ GOOD run of @SUMA_Make_Spec_FS" \
|& tee -a ${dir_echo}/o.01_suma_makespec_${subj}.txt
endif
# ===================================================================
JUMP_EXIT:
# ---------------- Biowulf slurm finish and copying ----------------
# Again, Biowulf-running considerations: if processing went fine and
# we were using a temporary directory, copy data back.
if( $usetemp && -d ${tempdir} ) then
echo "++ Copy from: ${tempdir}"
echo " to: ${dir_fs}"
\mkdir -p ${dir_fs}
\cp -pr ${tempdir}/${subj} ${dir_fs}/.
endif
# ----------------------------------------------------------------------
if ( $EXIT_CODE ) then
echo "** Something failed in Step ${EXIT_CODE} for subj: ${subj}"
else
echo "++ Copy complete for subj: ${subj}"
endif
# ===================================================================
Et voila!
Here is an example of the job info while running recon-all using a
script similar to the above (based on the job info, I moderated the
memory allocation above, to be more efficient).
Example Biowulf Dashboard job info |
|---|
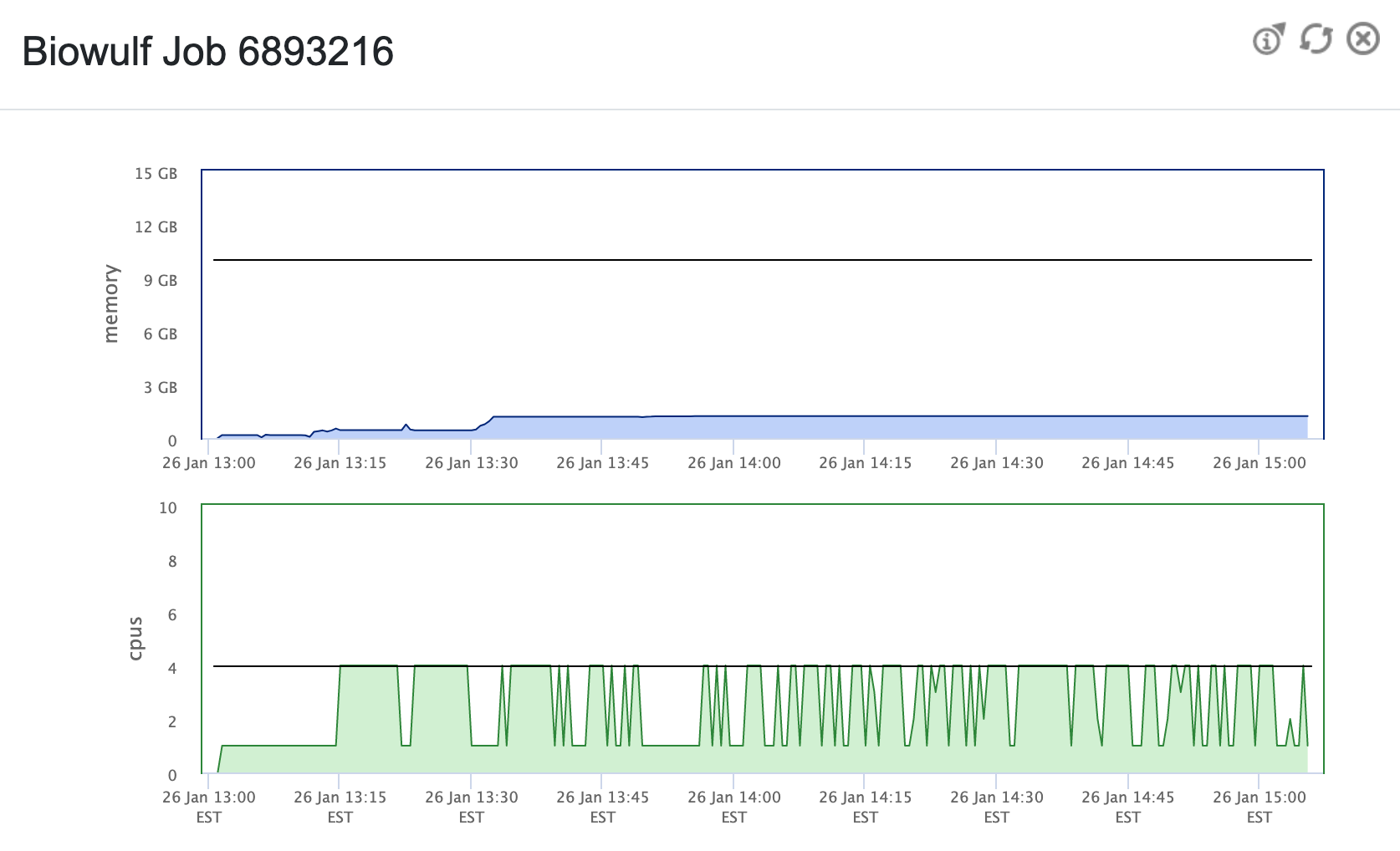
|
Looping over a group of subjects¶
The above could be made into a script used to loop over subjects by
having another script have a list of subject IDs and paths. That
script could loop over those subjects, submitting an sbatch job
every time, with this script taking 4 arguments (to fill the top
variables).
